



















In pharmaceutical, biotechnology, medical device, cell and gene therapy, and advanced manufacturing environments, the terms GMP and cGMP are often used interchangeably. But when it comes to cleanroom design, construction, qualification, and long-term performance, the distinction matters.
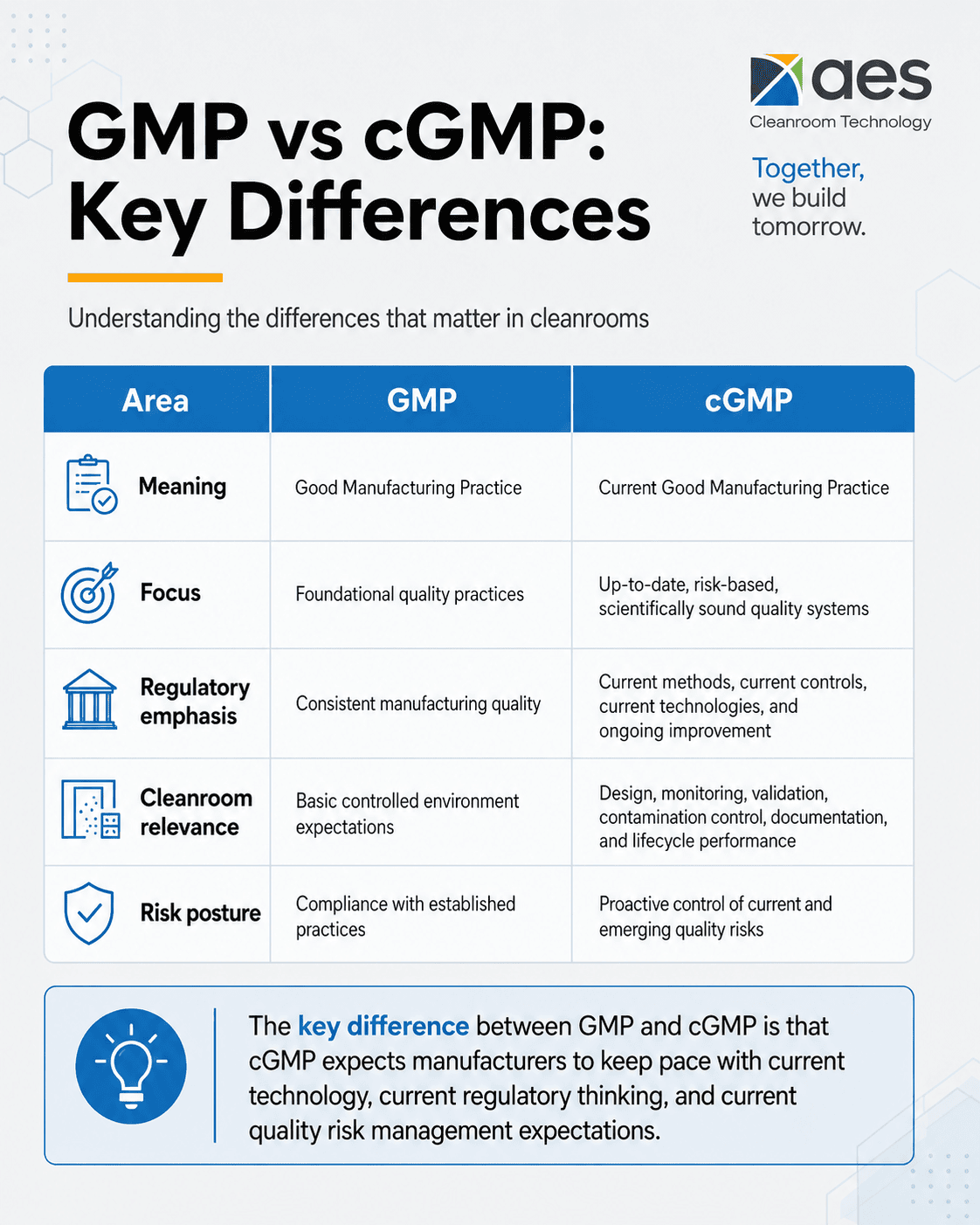
At a high level, GMP stands for Good Manufacturing Practice, while cGMP stands for current Good Manufacturing Practice. That single word – current – carries major implications for cleanroom owners, quality teams, facility planners, and manufacturers operating in regulated environments.
The FDA describes cGMP as the regulatory standard that provides systems for the proper design, monitoring, and control of manufacturing processes and facilities. Adherence to cGMP helps assure the identity, strength, quality, and purity of drug products by requiring manufacturers to adequately control manufacturing operations.
For cleanrooms, the difference between GMP and cGMP is not just a terminology issue. It affects how facilities are designed, how materials and personnel move through the space, how contamination risks are controlled, how systems are documented, and how a facility remains inspection-ready over time.
For organizations planning or upgrading modular cleanroom solutions, understanding cGMP vs GMP early can help align the facility with current regulatory expectations, operational needs, contamination control goals, and long-term scalability.
What Does cGMP Stand For?
cGMP stands for current Good Manufacturing Practice.
The “c” means current, which emphasizes that manufacturers are expected to use up-to-date systems, technologies, controls, procedures, and quality practices that reflect modern regulatory expectations. According to the FDA, cGMP requirements are flexible so manufacturers can use scientifically sound design, processing methods, testing procedures, modern technologies, and innovative approaches to achieve higher quality through continual improvement.
In other words, cGMP is not a static checklist. It is a quality framework that evolves with better science, improved cleanroom technologies, stronger contamination control strategies, improved monitoring systems, and more advanced documentation practices.
That is why early conceptual facility planning is so important. When cleanroom classifications, GMP zoning strategies, airflow needs, personnel flows, and process requirements are considered early, the facility can be designed around the realities of the manufacturing process rather than retrofitted around compliance gaps later.
cGMP Meaning in Cleanroom Environments
The cGMP meaning becomes especially important in cleanrooms because cleanrooms are not simply controlled spaces. In regulated life science environments, they are part of the manufacturing quality system.
A cleanroom that supports cGMP manufacturing must be designed and operated to control risks that can affect product quality, patient safety, and regulatory compliance. This can include:
- Airborne particulate control
- Microbial contamination control
- Personnel and material flow
- Pressure cascades
- HVAC performance
- Cleanable surfaces
- Gowning procedures
- Environmental monitoring
- Equipment integration
- Cleaning and disinfection
- Documentation and data integrity
- Qualification, validation, and ongoing maintenance
FDA’s cGMP regulations for drugs contain minimum requirements for the methods, facilities, and controls used in manufacturing, processing, and packing drug products. These requirements are intended to help ensure that a product is safe for use and has the ingredients and strength it claims to have.
For cleanroom projects, this means the facility itself must support the manufacturing process, the quality system, and the contamination control strategy – not just meet a particle count on the day of certification.
This is especially important for BioPharma cleanroom environments where aseptic processing zones, pressure cascading, flexible layouts, and scalable manufacturing environments may be required to support complex production workflows.
GMP vs cGMP: What Is the Difference?
The simplest way to understand GMP vs cGMP is this:
GMP refers to Good Manufacturing Practice – the broad quality principles used to ensure products are consistently produced and controlled according to applicable quality standards.
cGMP refers to current Good Manufacturing Practice – the current, enforceable regulatory expectation that manufacturers use modern, scientifically sound, and appropriately controlled systems to meet quality requirements.
In the United States, FDA regulations use the term current good manufacturing practice. Under 21 CFR Part 210, cGMP regulations establish minimum requirements for the methods, facilities, and controls used in manufacturing, processing, packing, or holding drugs to help ensure safety, identity, strength, quality, and purity.
Key Difference Between GMP and cGMP
The key difference between GMP and cGMP is that cGMP expects manufacturers to keep pace with current technology, current regulatory thinking, and current quality risk management expectations.
For cleanrooms, that expectation should influence the entire project lifecycle – from early planning and cleanroom design-build strategy to installation, qualification, operation, maintenance, and future expansion.
Why the “Current” in cGMP Matters for Cleanrooms
The word “current” is critical because cleanroom expectations evolve. Facility designs, monitoring systems, cleanroom materials, HVAC strategies, barrier technologies, and contamination control expectations have changed significantly over time.
A cleanroom built to an outdated standard may still look clean, but that does not mean it supports current regulatory expectations. cGMP cleanrooms must be designed, built, qualified, maintained, and documented in a way that reflects the current needs of the process and the current risks to product quality.
This is especially important for pharmaceutical manufacturing, biologics, cell and gene therapy, sterile manufacturing, advanced therapies, and other life science applications where contamination risks can directly affect product safety.
EU GMP Annex 1 emphasizes contamination control strategy, cleanroom classification, qualification, validation, monitoring, personnel gowning, and quality risk management for sterile manufacturing environments. It also notes that some Annex 1 principles may be useful for non-sterile products where microbial, particulate, or endotoxin/pyrogen control is important.
For teams developing or scaling therapies, AES’s Life Science cleanroom solutions are designed to support the speed, cleanliness, control, and compliance needs of evolving life science operations.
Cleanroom ISO Classification Is Not the Same as cGMP Compliance
A common misconception is that an ISO-classified cleanroom is automatically GMP- or cGMP-compliant. It is not.
ISO 14644-1 specifies the classification of air cleanliness by concentration of airborne particles in cleanrooms and clean zones. It focuses on particle concentration within defined particle-size ranges and does not characterize the physical, chemical, radiological, viable, or biological nature of airborne particles.
That distinction matters.
An ISO 7 or ISO 8 cleanroom classification may help define airborne particle cleanliness, but cGMP compliance involves much more than the ISO class. A cGMP cleanroom must also support the intended manufacturing process, contamination control strategy, personnel practices, documentation requirements, cleaning protocols, pressure relationships, equipment layout, maintenance access, and quality oversight.
In other words, ISO classification is a cleanroom performance metric. cGMP is a manufacturing quality expectation.
For teams still working through cleanroom terminology, layouts, classifications, and modular construction considerations, AES’s cleanroom FAQ resource can provide additional context.
What Makes a Cleanroom cGMP-Ready?
A cGMP-ready cleanroom is planned around the process, product, regulatory expectations, and lifecycle needs of the facility. For AES Cleanroom Technology, this is where integrated cleanroom design, modular cleanroom construction, HVAC coordination, and long-term serviceability become central to project success.
AES’s Faciliflex® turnkey modular cleanroom service supports modular cleanroom delivery from planning through execution, helping teams accelerate project timelines while maintaining control over critical cleanroom performance considerations.
1. Facility Design That Supports the Process
cGMP cleanroom design should begin with the manufacturing process, not just the room classification. Important planning questions include:
- What product is being manufactured?
- Is the process sterile, aseptic, low bioburden, or non-sterile?
- What are the critical contamination risks?
- Where do personnel, materials, equipment, and waste move?
- What pressure relationships are required?
- What utilities must be integrated?
- What future expansion or process change is likely?
The facility layout should reduce unnecessary movement, prevent cross-contamination, support efficient cleaning, and make the process easier to control.
That is why AES Compass™ can be valuable during early project planning. AES Compass™ supports conceptual facility planning by helping teams evaluate cleanroom classifications, GMP zoning strategies, airflow needs, process requirements, and operational flow before detailed design and construction begin.
2. Contamination Control Strategy
A cGMP cleanroom should support a defined contamination control strategy. This includes controlling viable and non-viable contamination risks through facility design, airflow, filtration, cleaning, disinfection, gowning, material transfer, process segregation, and monitoring.
For sterile manufacturing, EU GMP Annex 1 emphasizes that facilities, equipment, and processes should be appropriately designed, qualified, validated, and subject to ongoing verification. It also notes that technologies such as RABS, isolators, robotics, rapid methods, and continuous monitoring systems should be considered where appropriate to increase product protection.
Cleanroom construction materials also influence contamination control. AES’s resource on modular cleanroom cleaning and disinfection explores how cleanroom design, construction techniques, and materials can affect cleaning, disinfection, schedule, quality, and lifecycle cost.
3. Cleanable, Durable Materials
Cleanroom materials must support the cleaning and disinfection program. This makes walls, ceilings, doors, windows, floors, corners, penetrations, and transitions especially important.
In cGMP environments, materials should be selected to reduce contamination harborage points, withstand cleaning agents, support proper sealing, and maintain performance over time.
AES’s modular cleanroom walls and ceilings serve as foundational components for modular cleanroom environments, supporting retrofits, expansions, and new construction with cleanroom-ready finishes and controlled manufacturing consistency.
4. HVAC and Pressure Control
Cleanroom HVAC is not just about comfort. It plays a direct role in contamination control, room recovery, temperature and humidity management, airflow direction, pressure cascades, and product protection.
A cGMP-focused HVAC strategy should align with the room classification, process risk, equipment loads, personnel loads, and operational state of the facility.
For complex life science and BioPharma projects, integrated delivery matters. AES provides turnkey cleanrooms with integrated HVAC designed to streamline projects from concept to completion.
5. Qualification, Validation, and Documentation
A cleanroom must be more than well-built. It must be documented and verified.
cGMP environments typically require documented evidence that the cleanroom and supporting systems perform as intended. This can include design qualification, installation qualification, operational qualification, performance qualification, environmental monitoring, cleaning validation support, maintenance records, and change control documentation.
Data integrity is also a cGMP concern. FDA guidance explains that data integrity is central to cGMP for drugs and that FDA expects data to be reliable and accurate.
For organizations evaluating the experience and execution history of a cleanroom partner, reviewing completed cleanroom projects can help connect design-build claims to real-world facility outcomes.
6. Lifecycle Maintenance and Change Control
cGMP does not end after installation or certification. Cleanroom performance must be maintained over time.
That means service, inspection, filter replacement, seal checks, airflow verification, pressure monitoring, calibration, preventive maintenance, and documented corrective actions are all part of keeping the facility aligned with current expectations.
A proactive cleanroom service and maintenance program can help protect cleanroom performance, support ongoing facility reliability, and reduce the risk of avoidable downtime or compliance issues.
Why cGMP vs GMP Matters During Cleanroom Planning
Understanding cGMP vs GMP early in the project helps avoid costly mistakes later.
A cleanroom that is planned only around baseline GMP expectations may miss key design details that affect future compliance, such as:
- Insufficient space for personnel/material flows
- Poorly placed doors or transfer points
- Inadequate pressure cascade strategy
- Difficult-to-clean surfaces or transitions
- HVAC systems that cannot support process loads
- Poor maintenance access
- Insufficient documentation or turnover packages
- Lack of flexibility for future process changes
When cGMP expectations are built into the project from the beginning, teams can better align the cleanroom with regulatory needs, operational efficiency, contamination control, and long-term facility performance.
This is one reason off-site manufacturing and modular systems can be valuable for regulated environments. AES’s off-site cleanroom manufacturing approach supports faster installation, reduced site disruption, and quality control by fabricating modular cleanroom components in a controlled manufacturing environment.
Common Misunderstandings About GMP vs cGMP
Misunderstanding 1: “GMP and cGMP mean the same thing.”
They are closely related, but not identical. cGMP emphasizes the current state of good manufacturing practice, including current technologies, current controls, and current quality expectations.
Misunderstanding 2: “An ISO-classified cleanroom is automatically cGMP-compliant.”
ISO classification is important, but it does not equal cGMP compliance. ISO 14644-1 focuses on airborne particle concentration, while cGMP includes broader manufacturing controls, documentation, quality systems, and contamination control expectations.
Misunderstanding 3: “cGMP only applies to operations, not facility design.”
Facility design is a major part of cGMP readiness. FDA describes cGMP as including proper design, monitoring, and control of manufacturing processes and facilities.
Misunderstanding 4: “Once the cleanroom is built, cGMP compliance is complete.”
cGMP is ongoing. Cleanrooms must be maintained, monitored, documented, and improved as processes, technologies, and regulatory expectations evolve.
The Bottom Line: cGMP Cleanrooms Require Current Thinking
The difference between GMP and cGMP comes down to expectations. GMP establishes the foundation. cGMP raises the bar by requiring that facilities, processes, systems, controls, and documentation remain current.
For cleanrooms, that means cGMP readiness should influence every phase of the project:
- Conceptual planning
- Cleanroom layout
- HVAC design
- Material selection
- Personnel and material flow
- Contamination control
- Qualification and validation
- Documentation
- Maintenance
- Future scalability
A cleanroom designed with cGMP expectations in mind is not only cleaner – it is more defensible, more adaptable, more efficient, and better aligned with the realities of regulated manufacturing.
For life science organizations planning a new cleanroom, expanding an existing facility, or upgrading an aging controlled environment, the most important question is not simply, “What ISO class do we need?”
The better question is:
How do we design a cleanroom that supports current regulatory expectations, current contamination control strategies, and current manufacturing goals – today and into the future?
For teams preparing a new facility or evaluating cleanroom upgrades, contact AES Cleanroom Technology to discuss modular cleanroom design, manufacturing, construction, maintenance, or service needs.
FAQ: cGMP vs GMP in Cleanrooms
What does cGMP stand for?
cGMP stands for current Good Manufacturing Practice. The “c” stands for “current,” which means manufacturers are expected to use up-to-date systems, technologies, procedures, and controls that reflect modern quality and regulatory expectations.
What is the cGMP meaning in cleanrooms?
In cleanrooms, cGMP meaning refers to the current good manufacturing practices needed to control contamination, protect product quality, support regulatory compliance, and maintain documented control over the cleanroom environment. It includes facility design, HVAC performance, personnel flow, material flow, cleaning, monitoring, qualification, documentation, and ongoing maintenance.
What is the difference between GMP and cGMP?
The main difference between GMP and cGMP is that GMP refers to general Good Manufacturing Practice, while cGMP refers to current Good Manufacturing Practice. cGMP emphasizes up-to-date methods, current technology, modern quality systems, and continuous improvement.
Is cGMP better than GMP?
cGMP is not simply “better” than GMP – it is the current regulatory expectation in FDA-regulated drug manufacturing. cGMP builds on GMP principles by requiring manufacturers to keep their facilities, controls, systems, and procedures aligned with current standards and technologies.
Why is cGMP important in cleanroom design?
cGMP is important in cleanroom design because the cleanroom is part of the manufacturing control strategy. A cGMP cleanroom must support contamination control, process flow, cleaning, environmental monitoring, personnel practices, documentation, and long-term quality performance.
Does an ISO-classified cleanroom meet cGMP requirements?
Not automatically. ISO classification measures airborne particle cleanliness under ISO 14644-1, but cGMP includes broader requirements around manufacturing controls, documentation, facility design, contamination control, quality systems, and ongoing monitoring.
What is the difference between cGMP vs GMP for pharmaceutical cleanrooms?
For pharmaceutical cleanrooms, cGMP vs GMP usually comes down to whether the facility reflects current FDA expectations for manufacturing quality. cGMP cleanrooms must support current contamination control practices, modern monitoring, proper documentation, validated systems, and ongoing control of the manufacturing environment.
Why does the “current” in cGMP matter?
The “current” in cGMP matters because technology, regulatory expectations, contamination control practices, and manufacturing risks evolve. A cleanroom that was acceptable years ago may need upgrades or procedural improvements to remain aligned with current expectations.
What cleanroom features support cGMP readiness?
cGMP-ready cleanrooms often include cleanable wall and ceiling systems, properly designed HVAC, controlled pressure relationships, personnel and material flow controls, appropriate gowning areas, environmental monitoring points, sealed penetrations, durable surfaces, documented qualification, and ongoing maintenance programs.
Who needs a cGMP cleanroom?
Organizations manufacturing or supporting regulated life science products may need a cGMP cleanroom, especially in pharmaceutical, biotechnology, biologics, cell and gene therapy, sterile manufacturing, medical device, and advanced therapy applications.
Sources
- FDA – Facts About the Current Good Manufacturing Practice
- FDA – Current Good Manufacturing Practice Regulations
- eCFR – 21 CFR Part 210: Current Good Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs
- eCFR – 21 CFR Part 211: Current Good Manufacturing Practice for Finished Pharmaceuticals
- ISO – ISO 14644-1:2015 Cleanrooms and Associated Controlled Environments
- European Commission – EU GMP Annex 1: Manufacture of Sterile Medicinal Products
- FDA – Data Integrity and Compliance With Drug cGMP: Questions and Answers

